Case 1: Small Molecule Pure Substance (Benzene)
Case 1: Small Molecule Pure Substance (Benzene)
Section titled “Case 1: Small Molecule Pure Substance (Benzene)”In this case, we use benzene as a representative example of a small molecule pure substance. We will explain the basic flow of creating a molecule object from SMILES, packing it into a unit cell at a specified density, and performing structural relaxation.
Target System
Section titled “Target System”- Compound: Benzene
- Composition: 150 molecules (Total atoms: 1,800)
- Initial Target Density: 0.80 g/cm³
Calculation Code (run.py)
Section titled “Calculation Code (run.py)”import fbtk
# 1. Create a molecule object from SMILESbenzene = fbtk.Molecule.from_smiles("c1ccccc1", name="benzene")
# 2. Initialize the Builderbuilder = fbtk.Builder(density=0.80)
# 3. Add molecules to the systembuilder.add_molecule(benzene, count=150)
# 4. Build the systemsystem = builder.build()
# 5. Execute structural relaxation (Pre-relaxation)print("Starting relaxation for Benzene...")system.relax()
# 6. Output resultsbenzene.to_file("benzene.mol") # Individual moleculesystem.to_file("benzene_cell.mol2") # Relaxed unit cellprint("Done.")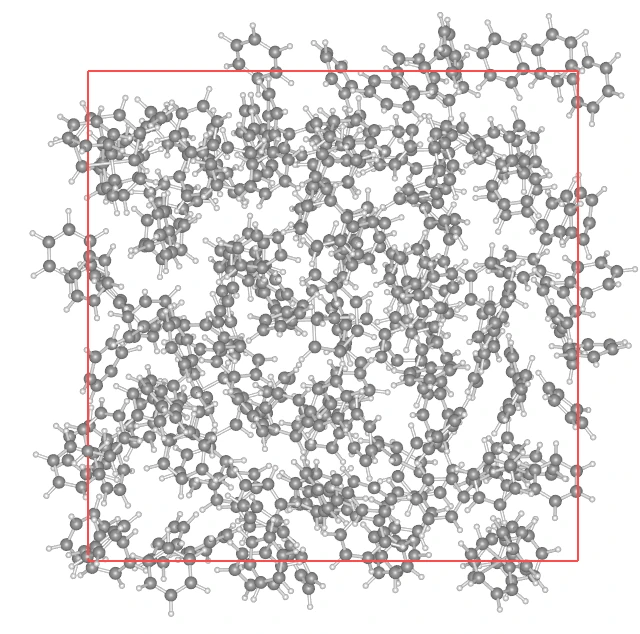
Reviewing the Execution Log
Section titled “Reviewing the Execution Log”During execution, the standard output displays the progress of the optimization process.
Atoms: 1800 | Bonds: 1800-------------------------------------------------------------------------------- | Fmax | FRMS | Total E |Iter | (kcal/mol/Å) | (kcal/mol/Å) | (kcal/mol) | Status-------------------------------------------------------------------------------- 0 | 36968.7497 | 6300.0166 | 686143734596.2736 | 10 | 4785.6728 | 229.2529 | 156059.5598 | 20 | 2074.8591 | 129.3691 | 87049.2019 | ... 980 | 2.0817 | 0.1992 | 18859.4087 | 990 | 36.2483 | 1.3258 | 18848.6857 |--------------------------------------------------------------------------------=== Optimization Finished ===Reason: Max-IterTotal Time: 14.335s (Avg: 14.335ms / step)Final Energy: 18852.1265 kcal/molFinal Fmax: 2.3571 kcal/mol/ÅFinal FRMS: 0.2088 kcal/mol/ÅMin Distance: 1.0809 Å (Atoms 553 and 559)--------------------------------------------------------------------------------Result Explanation
Section titled “Result Explanation”Effect of Structural Relaxation
Section titled “Effect of Structural Relaxation”If the Min Distance (closest interatomic distance) after relaxation is secured at 1.0 Å or more, it can be said that local “atomic collisions” have been resolved and a physically reasonable initial structure has been obtained.
Integration with ASE
Section titled “Integration with ASE”By using the to_ase() method, you can convert the system to an ASE Atoms object, allowing for easy integration with external MD software and various Calculators.
atoms = system.to_ase()# atoms.calc = SomeCalculator(...)